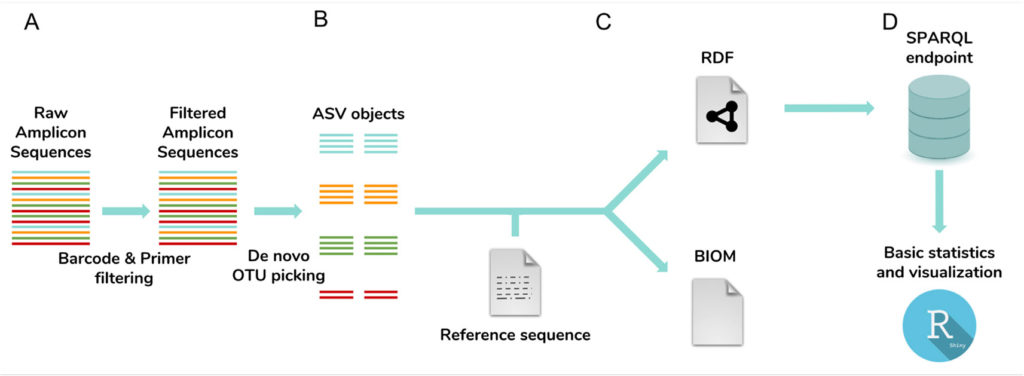
As a working example, we used existing raw 16S rRNA gene data from the DIABIMMUNE Microbiome project and NG-Tax 2.0 for data analyses. To this end, we downloaded raw amplicon data of over 1800 microbial samples from the project. Next, the data was automatically ingested by the UNLOCK infrastructure and stored according to the ISA standard. We also captured the metadata of these samples. Then, NG-Tax 20 automatically analysed the amplicon data to generate ASVs as we described above and exemplified below. The queries used to generate the examples shown can also be embedded in or be part of standard operating procedures (SOPs). Further post-processing can take place using structured data analysis processes integrated in Jupyter Notebooks.
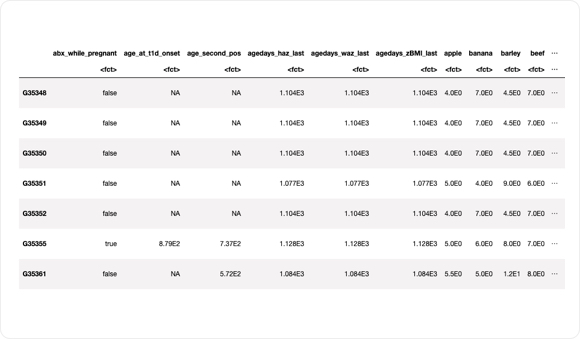
As the table above shows, users can directly sort and access metadata through the semantic framework. This enables users to easily select samples of interest for further downstream analysis.
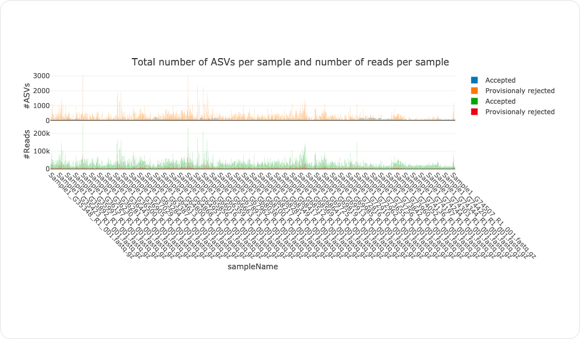
As shown in the graph above, amplicon data is noisy. NG-Tax 2.0, however, suppresses this noise by provisionally rejecting ASVs of low abundance. Moreover, due to the extensive incorporation of metadata and sequence information, users can access and study each (potentially rejected) ASV individually.
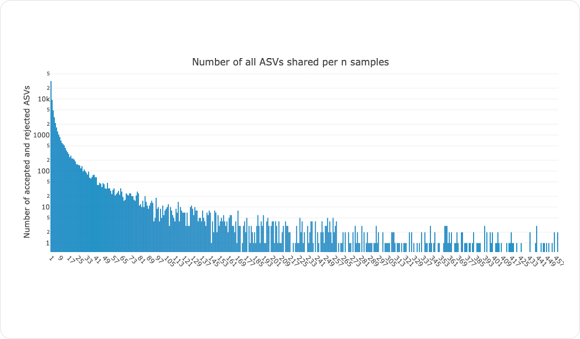
Unlike OTUs obtained by clustering of nearly identical sequences, Amplicon Sequence Variants suppoedly better represent a single species. Since NG-Tax 2.0 extracts ASVs and stores them in a semantic database, it enables cross-mapping of ASVs between thousands of samples. As shown in the grah above, we have plotted the number of ASVs, including the provisionally rejected ASV shared between samples.
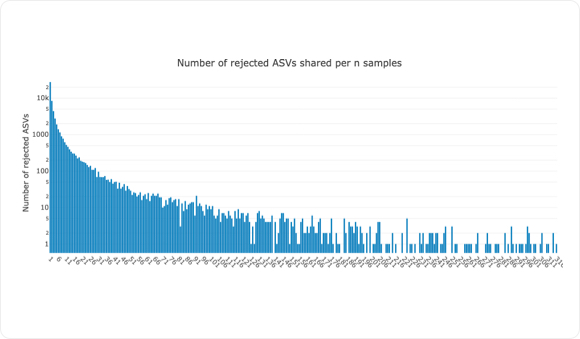
Next, within each sample, NG-Tax analyses and potentially flags ASVs as rejected due to severely low abundant reads, as seen in graph above. These ASVs might be falsely flagged, but through cross-mapping within and between samples these ASVs can become accepted and used for further downstream analysis.

For each individual sample, NG-Tax 2.0 provisionally rejects ASVs of low abundance. While most of these ASVs are likely random noise signals, a provisionally rejected ASV can also point at microbial species with a particular low abundance in the specific sample. We can study this by asking two related questions.
1. “Do we find a particular provisionally rejected ASV in more samples?”
2. “Is this ASV an accepted ASV in other samples?”
As the ASVs are stored in a graph database, this and the results presented in the examples above, can be done with standard SPARQL queries. In the example, we have plotted the number of the provisionally rejected ASV that are shared between the 1800 samples. As can be seen, by far the majority occurs in less than three samples and on the basis of this most probably most of them are noise ASvs. The example SPARQL query above explores this a little bit further and provides an answer to the question “How many provisionally rejected ASVs are in fact accepted ASVs in other samples?”.